Laser improves the time resolution of CryoEM

In 2017, Jacques Dubochet, Joachim Frank, and Richard Henderson won the Nobel Prize in Chemistry for their contributions to cryo-electron microscopy (cryoEM), an imaging technique that can capture pictures of biomolecules such as proteins with atomic precision.
In cryoEM, samples are embedded in vitreous ice, a glass-like form of ice that is obtained when water is frozen so rapidly that crystallization cannot occur. With the sample vitrified, high-resolution pictures of their molecular structure can be taken with an electron microscope, an instrument that forms images using a beam of electrons instead of light.
CryoEM has opened up new dimensions in life sciences, chemistry, and medicine. For example, it was recently used to map the structure of the SARS-CoV-2 spike protein, which is the target of many of the COVID-19 vaccines.
Proteins constantly change their 3D structure in the cell. These conformational rearrangements are integral for proteins to perform their specialized functions, and take place within millionths to thousandths of a second. Such fast movements are too fast to be observed in real time by current cryoEM protocols, rendering our understanding of proteins incomplete.
But a team of scientists led by Ulrich Lorenz at EPFL’s School of Basic Sciences has developed a cryoEM method that can capture images of protein movements at the microsecond (a millionth of a second) timescale. The work is published in Chemical Physics Letters.
The method involves rapidly melting the vitrified sample with a laser pulse. When the ice melts into a liquid, there is a tunable time window in which the protein can be induced to move in the way they do in their natural liquid state in the cell.
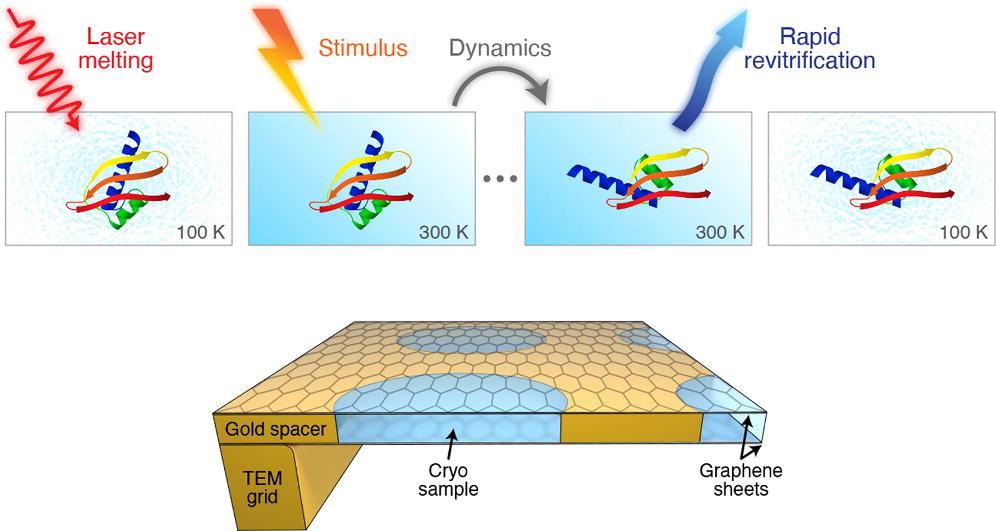
“Generally speaking, warming up a cryo sample causes it to de-vitrify,” says Ulrich Lorenz. “But we can overcome this obstacle by how quickly we melt the sample."
After the laser pulse, the sample is re-vitrified in just a few microseconds, trapping the particles in their transient configurations. In this “paused” state, they can now be observed with conventional cryoEM methods.
“Matching the time resolution of cryoEM to the natural timescale of proteins will allow us to directly study processes that were previously inaccessible,” says Lorenz.
The team of scientists tested their new method by disassembling proteins after structurally damaging them, and trapping them in partially unraveled configurations.
