Rezeptorproteinen beim Verbiegen zuschauen
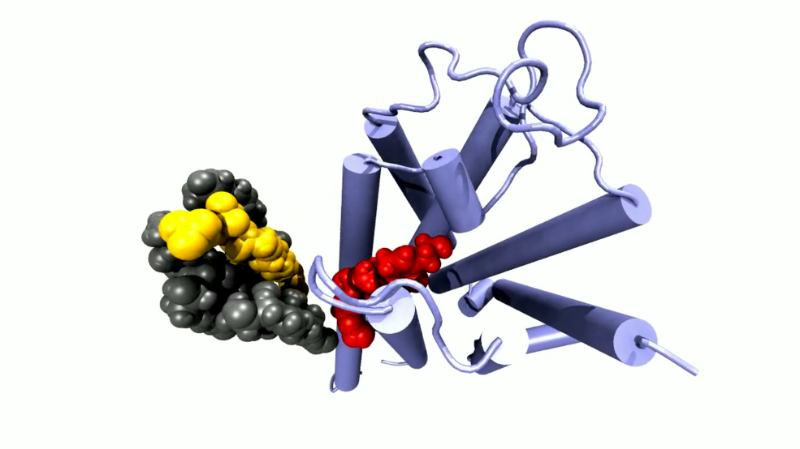
Herr Guixà-González, Sie erforschen seit vielen Jahren G-Protein-gekoppelte Rezeptoren. Warum sind die so interessant?
G-Protein-gekoppelte Rezeptoren sind schlichtweg überall – fast jede Zelle in unserem Körper braucht sie. Diese Proteinmoleküle ragen wie Antennen aus einer Zelle heraus. An sie docken beispielsweise Moleküle an, die im Blut schwimmen. Sie sind daher auch wichtige Ziele für Wirkstoffe in der Medizin. Man schätzt, dass über ein Drittel aller derzeit zugelassenen Medikamente ihre Wirkung über diese Familie von Proteinen vermittelt.
Wie genau agieren diese Rezeptoren?
Sie sind in die Zellmembran eingebettet und vermitteln Informationen ins Zellinnere. Nehmen wir an, man ist aufgeregt und der Körper hat Adrenalin ausgeschüttet, welches nun im Blut zirkuliert. Es bindet dann beispielsweise an den G-Protein-gekoppelten Rezeptor in der Membran einer Herzmuskelzelle. Daraufhin ändert der Rezeptor seine Form, er wird aktiviert, wie wir sagen. Im aktiven Zustand kann der Rezeptor Proteine im Inneren der Zelle binden und aktivieren. Wenn er jetzt seine Form, vermittelt durch Adrenalin, ändert, werden diese Proteine freigesetzt und können eine Funktion ausüben. Im Falle der Herzmuskelzelle wird ein Enzym aktiviert, das über eine Kaskade dann am Ende dafür sorgt, dass das Herz schneller schlägt.
Und in anderen Zellen passiert vermutlich etwas anderes?
Richtig. Es gibt über 800 verschiedene Typen von G-Protein-gekoppelten Rezeptoren. Neben Adrenalin können sie eine Unmenge anderer Dinge registrieren: Hormone und Neurotransmitter im Gehirn etwa. Ebenso Sehen und Geschmack. Auch wenn wir etwas riechen, binden Moleküle an solche Rezeptoren.
Wie können Medikamente in diesen Prozess eingreifen?
Nehmen wir mal an, jemand hat eine Krankheit, bei der einer der Rezeptoren permanent aktiviert ist. Man entwickelt dann ein Medikament, das an den Rezeptor bindet und ihn blockiert. Oder jemand will umgekehrt ein Medikament entwickeln, das den Rezeptor aktiviert. In beiden Fällen brauchen wir ein Molekül, das genau zu dem Rezeptor passt.
Das ist dann ein bisschen wie ein Puzzlespiel, bei dem man zwei zueinander passende Teile sucht?
In etwa, ja. Allerdings geht es – anders als bei einem Puzzle – nicht nur um die statische Struktur des Rezeptors. In der Realität läuft ein ganzer Film ab. Denn wenn ein Molekül an den Rezeptor bindet, verändert der dadurch seine Form. Und auch ohne Aktivierung ist das Ganze ein sehr dynamisches System: Wechselwirkungen innerhalb des Proteins werden permanent gelöst und wieder gefestigt. Die Frage ist: Wie laufen diese Änderungen genau ab?
Sie wollen also den ganzen Film sehen und nicht nur einzelne Bilder. Wie gehen Sie dafür vor?
Es ist ein bisschen so wie in den Anfängen der Fotografie. Damals wussten die Menschen nicht, wie genau Pferde galoppieren: Haben sie zu irgendeinem Zeitpunkt alle vier Füsse auf dem Boden oder nicht? Mit blossem Auge konnte man das nicht sehen, weil die Pferde zu schnell waren. Videokameras gab es noch nicht. Um den Vorgang zu beobachten, kann man viele Fotos machen, hintereinanderlegen und die Lücken mit Computersimulationen füllen. Das Pferd ist unser Rezeptorprotein und die Fotos sind Kristallstrukturen.
Kristallstrukturen?
Ja, in der Strukturbiologie arbeitet man oft mit Kristallstrukturen: Man ermittelt den Aufbau eines Biomoleküls, indem man seine Kristalle mit Röntgenlicht beschiesst und aus dem entstehenden Muster auf die dreidimensionale Struktur zurückrechnet. Solche Kristallstrukturen sind im Grunde aber nur Schnappschüsse. Wir nehmen solche Kristallstrukturen, die Forschende etwa mit der Synchrotron Lichtquelle Schweiz SLS oder dem Freie-Elektronen-Röntgenlaser SwissFEL ermittelt haben, erschaffen darauf basierend ein Modell und schauen uns dann an, wie sich die Moleküle nach den Gesetzen der klassischen Physik bewegen. So erhalten wir unseren Film.
Wie lassen sich die Bewegungen der Rezeptoren berechnen?
Anhand der Kristallstrukturen erstellen wir ein dreidimensionales Atommodell des Systems. Es zeigt die Position jedes einzelnen Atoms innerhalb des Proteins. Zusätzlich enthält das Modell die Lipidmoleküle der Zellmembran sowie Wassermoleküle und Salzionen. So ein Modell enthält leicht 100’000 Atome oder mehr. Mit der Technik der Molekulardynamik betrachten wir dann die Wechselwirkungen zwischen einzelnen Atomen oder Teilen des Proteins: Wie gross sind die Abstände, die Winkel, die Energien, wie stark sind daher die Wechselwirkungen und welche räumlichen Bewegungen ergeben sich daraus? So beginnen die Atome quasi zu tanzen – ein sehr physikalischer Tanz nach den Regeln der klassischen Physik. Mit dieser Methode simulieren wir die Bewegungen des Proteins über mehrere Mikrosekunden. Diese Berechnungen dauern allerdings mehrere Wochen, und wir benötigen dafür Supercomputer.
Eine gewaltige Rechenleistung also. Aber übernehmen die Forschungen am SwissFEL nicht eben diese Aufgabe, also das Erstellen von Filmen von Molekülbewegungen?
Die Idee hinter einem Freie-Elektronen-Röntgenlaser ist ja Folgende: Statt nur ein einziges Bild von einem statischen Protein zu machen, macht man gleich mehrere Bilder in sehr kleinen zeitlichen Abständen, während sich das Protein bewegt. Setzt man diese Bilder – sprich Kristallstrukturen – hintereinander, sieht man tatsächlich, wie sich das Protein bewegt. Aber zum gegenwärtigen Zeitpunkt kann der SwissFEL die Methode der Molekulardynamik nicht ersetzen. Denn bislang lassen sich im SwissFEL nur bestimmte Proteine untersuchen, das sind meist solche, die mit Licht aktiviert werden. Die meisten Proteine werden allerdings durch andere Dinge aktiviert, beispielsweise durch Hormone oder andere Proteine. In solchen Fällen ist die Molekulardynamik am besten geeignet, um Proteinbewegungen mit atomarer Auflösung zu untersuchen. Im Moment jedenfalls.
Gemeinsam mit einem grossen europäischen Forschungskonsortium und unter Leitung von Jana Selent von der Universität Pompeu Fabra in Barcelona wurde die Online-Plattform gpcrmd.org ins Leben gerufen – kurz für «G protein coupled receptors molecular dynamics». Was war das Ziel?
In unserem Forschungsfeld erstellen viele von uns dynamische Simulationen von G-Protein-gekoppelten Rezeptoren. Mit der neuen Plattform wollen wir möglichst viele dieser Simulationen der Wissenschaft allgemein zugänglich machen. Forschende aus der ganzen Welt können ihre Simulationen nun miteinander teilen und die von anderen Gruppen analysieren. Das ist bisher einzigartig.
Wie viele Simulationen enthält die Plattform bisher?
Derzeit über 600. Damit decken wir 70 Prozent aller Familien von G-Protein-gekoppelten Rezeptoren ab. Wir sind gerade dabei, eine zweite Version der Plattform vorzubereiten. Diese soll dann für jede Familie mindestens eine Simulation enthalten.
Wie hilft die Plattform bei der Medikamentenentwicklung?
Beispielsweise kann man sich anschauen, wie unterschiedliche Moleküle an den Rezeptor binden – und so überprüfen, wie man seinen potenziellen Wirkstoff verbessern kann. Man kann sich auch ansehen, wie sich Wassermoleküle typischerweise um den Rezeptor verteilen und die Wirkung von gebundenen Molekülen beeinflussen. Oder man untersucht, aus welchen Richtungen ein Wirkstoff überhaupt an den Rezeptor gelangen kann. Die Plattform bietet unzählige Möglichkeiten – und das alles kostenlos!
Interview: Paul Scherrer Institut/Brigitte Osterath
